
চিত্র:Phylogenetic tree of coronaviruses.jpg
এর চেয়ে বেশি রেজোলিউশন লভ্য নয়।
Phylogenetic_tree_of_coronaviruses.jpg (৫৭১ × ৪৫১ পিক্সেল, ফাইলের আকার: ৭৫ কিলোবাইট, এমআইএমই ধরন: image/jpeg)
| এই ফাইলটি উইকিমিডিয়া কমন্স থেকে নেওয়া। সেখানের বর্ণনা পাতার বিস্তারিত নিম্নে দেখানো হলো। (সম্পাদনা)
|
{kind=link}
{kind=link}
সারাংশ
| বিবরণ |
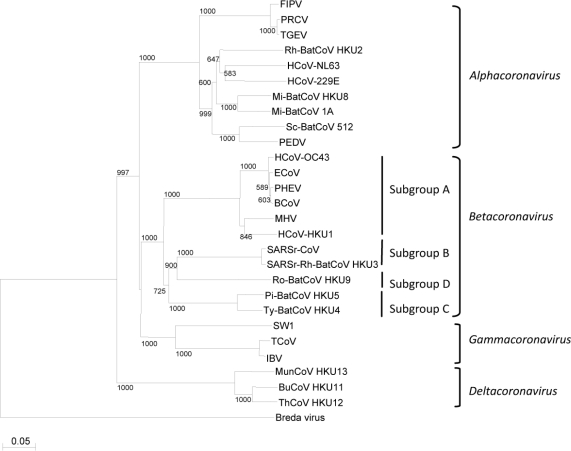
English: Phylogenetic analysis of RNA-dependent RNA polymerases (Pol) of coronaviruses with complete genome sequences available. The tree was constructed by the neighbor-joining method and rooted using Breda virus polyprotein. Bootstrap values were calculated from 1000 trees. 1118 amino acid positions in Pol were included. The scale bar indicates the estimated number of substitutions per 20 amino acids. All abbreviations for the coronaviruses were the same as those in Figure 1. |
| তারিখ | |
| উৎস | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ |
| লেখক | Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen |
লাইসেন্স প্রদান
এই ফাইলটি ক্রিয়েটিভ কমন্স অ্যাট্রিবিউশন ৩.০ আনপোর্টেড লাইসেন্সের আওতায় লাইসেন্সকৃত।
- আপনি স্বাধীনভাবে:
- বণ্টন করতে পারেন – এ কাজটি অনুলিপি, বিতরণ এবং প্রেরণ করতে পারেন
- পুনঃমিশ্রণ করতে পারেন – কাজটি অভিযোজন করতে পারেন
- নিম্নের শর্তাবলীর ভিত্তিতে:
- স্বীকৃতিপ্রদান – আপনাকে অবশ্যই যথাযথ স্বীকৃতি প্রদান করতে হবে, লাইসেন্সের একটি লিঙ্ক সরবরাহ করতে হবে এবং কোনো পরিবর্তন হয়েছে কিনা তা নির্দেশ করতে হবে। আপনি যেকোনো যুক্তিসঙ্গত পদ্ধতিতে এটি করতে পারেন। কিন্তু এমন ভাবে নয়, যাতে প্রকাশ পায় যে লাইসেন্সধারী আপনাকে বা আপনার এই ব্যবহারের জন্য অনুমোদন দিয়েছে।
ফাইলের ইতিহাস
যেকোনো তারিখ/সময়ে ক্লিক করে দেখুন ফাইলটি তখন কী অবস্থায় ছিল।
| তারিখ/সময় | সংক্ষেপচিত্র | মাত্রা | ব্যবহারকারী | মন্তব্য | |
|---|---|---|---|---|---|
| বর্তমান | ০২:৪২, ৭ মার্চ ২০২০ | | ৫৭১ × ৪৫১ (৭৫ কিলোবাইট) | Guest2625 | Uploaded a work by Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ with UploadWizard |
সংযোগসমূহ
নিচের পৃষ্ঠা(গুলো) থেকে এই ছবিতে সংযোগ আছে:
ফাইলের বৈশ্বিক ব্যবহার
নিচের অন্যান্য উইকিগুলো এই ফাইলটি ব্যবহার করে:
- en.wikipedia.org-এ ব্যবহার
- fa.wikipedia.org-এ ব্যবহার
- fr.wikipedia.org-এ ব্যবহার
- ig.wikipedia.org-এ ব্যবহার
- sq.wikipedia.org-এ ব্যবহার
- su.wikipedia.org-এ ব্যবহার
{kind=link}